Condel¶
Condel stands for CONsensus DELeteriousness score of non-synonymous single nucleotide variants (SNVs). The idea behind it is to integrate the output of computational tools aimed at assessing the impact of non synonymous SNVs on protein function. To do this, it computes a weighted average of the scores (WAS) of these tools. We originally developed Condel to integrate the outputs of five tools: SIFT, Polyphen2, MAPP, LogR Pfam E-value (implemented ad hoc following the instructions at Clifford RJ, Edmonson MN, Nguyen C, and Buetow KH (2004) Large-scale analysis of non-synonymous coding region single nucleotide polymorphisms. Bioinformatics 20, 1006-1014) and MutationAssessor.
We have now developed a new version of Condel which can be accessed using the FannsDB web server. This new version incorporates mainly three new features with respect to the original Condel web server (see below). While the first and second innovation affect the Condel score, the third one merely concerns the web server.
- The Condel score now contains up-to-date versions of the individual tools included. The mapping of the variants to genomic elements has also been updated to version 64 of the Ensembl database.
- The Condel score now consists in a weighted average of the scores of MutationAssessor and FatHMM. After exhaustive search of all possible combinations of weighted scores of SIFT, PolyPhen2, MutationAssessor and FatHMM, we found this combination to perform better than the others (see Performance figures below). Nevertheless, the scores of these five tools for your list of variants are still part of the results of Condel 2.0.
- Condel, SIFT, PolyPhen2, MutationAssessor, and FatHMM scores in FannsDB are now pre-computed for all possible variants in all human protein-coding genes. This greatly improves the performance of the FannsDB web server. You will retrieve the scores of your variants (regardless of their number) in a matter of seconds after you launch the query.
How does it work ?¶
As in the original Condel score, the scores of different methods are weighted using the complementary cumulative distributions of approximately 20.000 missense SNPs, both deleterious and neutral. The probability that a predicted deleterious mutation is not a false positive of the method and the probability that a predicted neutral mutation is not a false negative are employed as weights. For details, please read the Condel paper.
Performance¶
The development of new methods to identify likely deleterious missense variants (FatHMM in this case), together with the improvement of the existing ones, has improved the performance of Condel 2.0 (88.4%) with respect to the original version. Actually, the Condel 2.0, which includes only the scores of FatHMM and MutationAssessor exhibits the same accuracy as the original published tool, with five methods with scores computed by hand tuning the parameters of different methods to achieve the best results in the test dataset.
The figure below illustrates the performance metrics calculated for the different methods (including Condel).
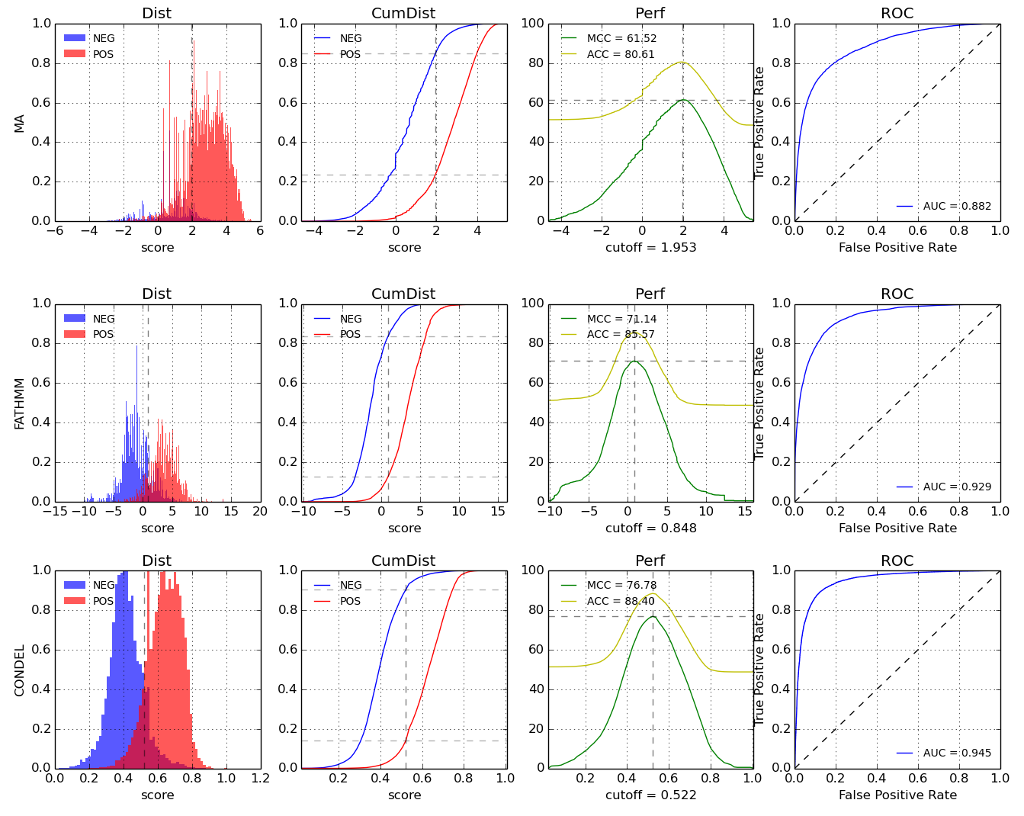
Performance metrics: There are 4 plots for each method (MutationAssessor, FatHMM and Condel), the first plot shows the density of scores for the known neutral (NEG) and deleterious (POS) mutations in HumVar. The scores of FatHMM are reversed so higher values represents more chance to deleteriouness. The second plot shows the cummulative probability distribution. The third plot shows the Matthews correlation coefficient (MCC) and the accuracy (ACC). The cutoff (marked with a vertical line) represents the threshold that maximizes the MCC. And the last plot represents the ROC curve. The numbers in the legend are the area under the curve (AUC).
The Figure below illustrates the ROC curves of the two individual methods and the Condel 2.0 on the aforementioned dataset. The Condel 2.0 score performs better than the two individual methods in the task of classifying mutations as deleterious or neutral.
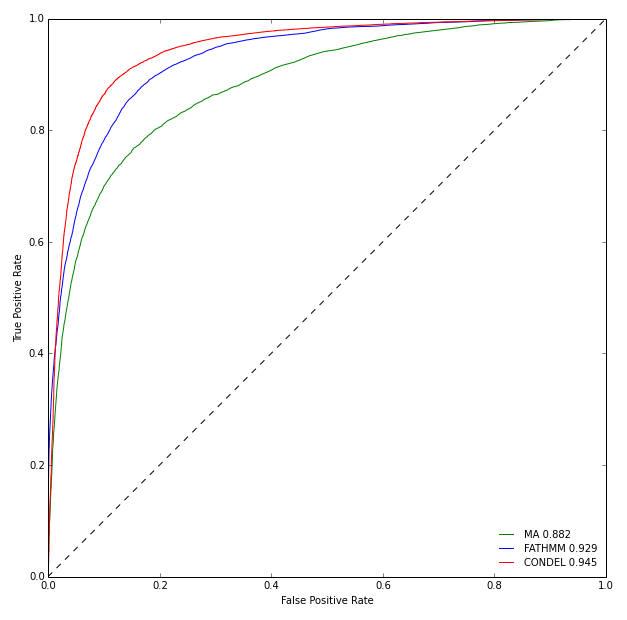
ROC curve of the individual methods and Condel: FatHMM, Mutation Assessor and Condel.
How to cite Condel¶
Please, cite our Condel paper:
Improving the Assessment of the Outcome of Nonsynonymous SNVs with a Consensus Deleteriousness Score, Condel (2011) Abel González-Pérez and Nuria López-Bigas, American Journal of Human Genetics 10.1016/j.ajhg.2011.03.004 Download PDF
Don’t forget to mention also the original tools:
- SIFT: PMID:19561590
- Polyphen2: PMID:20354512
- MutationAssessor: PMID:21727090
- FatHMM: PMID:23033316
- Ensembl-variation: PMID:20459805